导读部分由于其独特的电子,光学和化学性质,纳米材料被用于从化学品生产到医药和发光装置的各种应用中。但是当在其结构中引入另一种金属时,也
部分由于其独特的电子,光学和化学性质,纳米材料被用于从化学品生产到医药和发光装置的各种应用中。但是当在其结构中引入另一种金属时,也称为“掺杂”,研究人员不确定金属将占据哪个位置以及它将如何影响纳米团簇的整体稳定性,从而增加实验时间和成本。
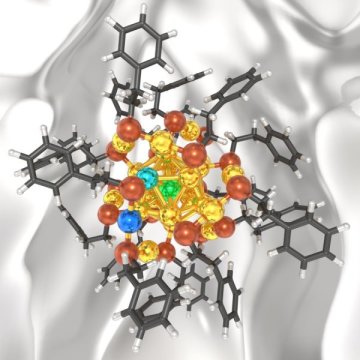
然而,匹兹堡大学斯旺森工程学院的研究人员开发了一种新理论,以更好地预测纳米团簇在给定金属被引入其结构时的行为方式。这项名为“配体保护金属纳米团簇的热力学稳定性”的研究报告刊登在ACS 物理化学快报的封面上。共同作者是Giannis Mpourmpakis,他是斯克森学院的二百周年校友教职研究员和化学与石油工程助理教授,以及博士候选人和NSF研究员Michael Taylor。他们的研究结果与先前的研究有关,这些研究主要集中在设
“设计纳米团簇的大小,形状和组成是控制其固有特性的一种方法,”Mpourmpakis博士说。“特别是,配体保护的Au(金)纳米团簇是一类纳米材料,其中已经实现了对其尺寸的精确控制。我们的研究旨在更好地预测其双金属对应物的形成方式,这将使我们能够更容易地预测它们的结构没有在实验室中进行过多的试验和错误实验。“
该研究在Mpourmpakis博士的计算机辅助纳米和能量实验室(CANELA。)中完成,使他们能够计算预测配体保护的Au纳米团簇中的确切掺杂剂位置和浓度。他们还发现,他们最近开发的理论,解释了实验合成的Au纳米团簇的确切尺寸,也适用于双金属纳米团簇,它具有更大的多功能性。
“这种计算理论现在可用于加速纳米材料的发现并更好地指导实验工作,”Mpourmpakis博士说。“更重要的是,通过在双金属纳米团簇上测试这一理论,我们有可能开发出具有定制特性的材料。这可能对纳米技术产生巨大影响。”
免责声明:本文由用户上传,如有侵权请联系删除!
猜你喜欢
最新文章










- 惠普今日发布了该公司的2022财年第三财季财报

2022-08-31 15:13:25
- MyFitnessPal将其流行的条形码扫描仪功能放在付费墙后面

2022-08-31 09:50:57
- 摩托罗拉系统收购无线电专家巴雷特

2022-08-30 14:12:44
- 安全hold公布2022年第二季度业绩

2022-08-29 14:47:40
- Wheels Up宣布创纪录的第二季度收入同比增长49%

2022-08-29 14:11:27